
Anemia mesdhetare (ose beta-talasemia) është një çrregullim i trashëguar i gjakut.
Pacientët që janë të prekur kanë më pak qeliza të kuqe të gjakut se normalja, me defekte në sintezën e hemoglobinës (Hb, proteina përgjegjëse për transportin e oksigjenit).
Anemia mesdhetare Më konkretisht, anemia mesdhetare është për shkak të prodhimit të ndryshuar të një ose më shumë prej katër zinxhirëve proteinikë (globinat) që përbëjnë Hb. Kjo rezulton në mungesë të oksigjenit në trup.
Entiteti i shqetësimit, simptomat dhe pasojat janë shumë të ndryshueshme dhe varen rrënjësisht nga lloji i defektit gjenetik. Në fakt, ekzistojnë 3 forma të ndryshme të anemisë mesdhetare:
 Talasemia major (ose sëmundja e Cooley);
Talasemia major (ose sëmundja e Cooley);
Talasemia intermedia;
Talasemia minore.
Në rastet më të rënda, anemia mesdhetare është invaliduese dhe vë në rrezik jetën e njerëzve; në forma të tjera, është pothuajse asimptomatike. Ekziston gjithashtu mundësia për të qenë një bartës i shëndetshëm, me rrezikun e gjenerimit të fëmijëve që do të zhvillojnë sëmundjen.
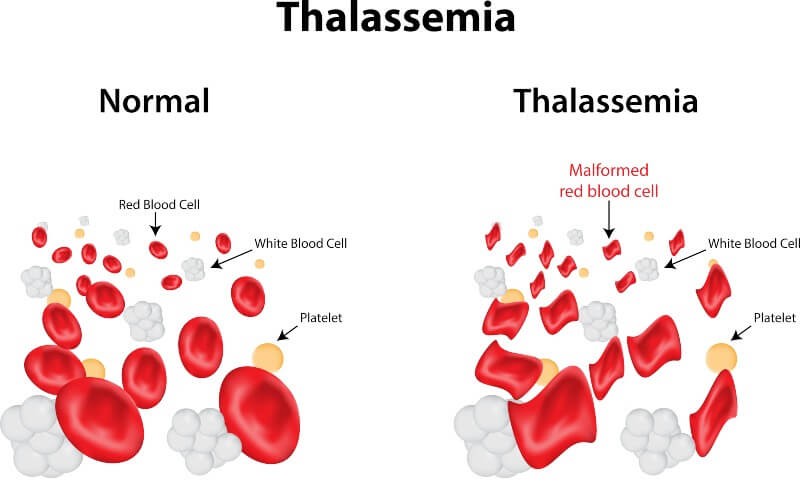
Anemia mesdhetare është e identifikueshme nga analizat gjenetike dhe analizat e gjakut; kjo e fundit do të tregojë praninë e rruazave të kuqe të gjakut me përmasa të çrregullta, të brishta, të pakta dhe më të vogla se normalja.
Trajtimi përfshin disa qasje, duke përfshirë pak a shumë transfuzione gjaku të përsëritura të lidhura me terapinë e kelimit (për të shmangur akumulimin e hekurit) dhe transplantin e palcës kockore nga donatorë të pajtueshëm. Ndonjëherë nuk nevojitet asnjë ndërhyrje terapeutike.
A ke ditur atë…
Anemia mesdhetare i detyrohet emrit të saj për faktin se fillimisht u vërejt në popullatat mesdhetare. Në Evropë, sëmundja është e përqendruar kryesisht në Greqi, brigjet turke dhe ishujt, duke përfshirë Siçilinë dhe Sardenjen.
Në territorin tonë kombëtar, zonat e deltës së Po dhe Ciociaria janë gjithashtu ndër vendet më të prekura. Vlerësohet se në Itali janë rreth 2.5 milionë njerëz që vuajnë nga anemia mesdhetare, edhe nëse shumica e tyre janë bartës të shëndetshëm.
Sëmundja është gjithashtu e përhapur në Azinë Qendrore dhe Juglindore, Indi dhe Kinë.
Anemia mesdhetare është një formë e talasemisë e shkaktuar nga një sintezë e reduktuar ose e munguar e zinxhirëve beta-globin të hemoglobinës.
Pasojat e këtij mosfunksionimi janë:
Pamja klinike e anemisë mikrocitike dhe hipokromike;
Shkatërrimi i hershëm i rruazave të kuqe të gjakut (hemoliza), me efekte të ndryshme në të gjithë organizmin;
Eritropoieza joefektive me ashpërsi të ndryshme (palca e eshtrave prodhon qeliza të kuqe të gjakut të vogla, të brishta dhe kryesisht të varfëra me oksigjen për t’u transportuar në inde).
Roli i hemoglobinës
Hemoglobina (Hb) është një proteinë e përfshirë në qelizat e kuqe të gjakut, e specializuar në transportin e oksigjenit në pjesë të ndryshme të trupit. Në një të rritur të shëndetshëm, përqendrimi i tij nuk duhet të bjerë nën 12 g/dl. Reduktimi i hemoglobinës, i shoqëruar me atë të rruazave të kuqe të gjakut në gjak, çon në simptoma që karakterizojnë aneminë mesdhetare.
Shkaqet
Anemia mesdhetare është një sëmundje hematologjike me origjinë gjenetike.
Kuadri klinike është për shkak të ndryshimeve shumë heterogjene (mutacione pikash, delecione, etj.) në gjenin HBB (11p15.5), i cili kodon beta-globinat thelbësore në sintezën e hemoglobinës. Duhet mbajtur mend, në fakt, se kjo molekulë përbëhet nga katër nënnjësi: dy zinxhirë alfa dhe dy zinxhirë beta.
Në bazën e anemisë mesdhetare ka një shmangie gjenetike që përfshin prodhimin e pakët (β+) ose mungesën (β0) të zinxhirëve beta: hemoglobina që rezulton është e pakët dhe dëmton membranën e qelizave të kuqe të gjakut, të cilat shkatërrohen.
Klasifikimi
Ekzistojnë 3 forma të ndryshme të anemisë mesdhetare, të klasifikuara sipas ashpërsisë së çrregullimit:
Talasemia major (ose sëmundja e Cooley): në përgjithësi shfaqet brenda moshës 2 vjeçare dhe, për të mbijetuar, kërkon transfuzion gjaku periodik gjatë gjithë jetës, përveç përdorimit të barnave specifike.
Talasemia intermedia: formë më pak e rëndë; mund të mbetet latente ose të manifestohet në mënyrë të ngjashme me atë kryesore dhe të kërkojë transfuzione të herëpashershme për të përmirësuar cilësinë e jetës.
Talasemia minore: përgjithësisht asimptomatike, nuk kërkon trajtime të veçanta, por kontrolle të rregullta të vlerave të gjakut.
Mekanizmat kryesorë gjenetikë përgjegjës për format e anemisë mesdhetare
Lloji i mutacionit
fenotip
Mutacione pikash të pakuptimta
β0-talasemia
Mutacionet e ekzoneve/introneve ose vendeve afër këtyre sekuencave, me ndryshime të procesit normal të formimit të mRNA
β+ dhe β0-talasemia
Mutacionet e sekuencave promotore
β+-talasemia
Fshirje të gjeneve të globinës (të rralla)
β0-talasemia
Si transmetohet
Transmetimi është autosomik recesiv, kështu që vetëm një fëmijë, prindërit e të cilit janë të dy bartës, mund të preket nga anemia mesdhetare.
Bartës i shëndetshëm i anemisë mesdhetare: cilat janë rreziqet?
Bartës të shëndetshëm të anemisë mesdhetare janë njerëzit që kanë defektin gjenetik që qëndron në themel të sëmundjes, por janë asimptomatikë dhe bëjnë një jetë normale.
Megjithatë, kjo gjendje mbart rreziqe. Nëse partneri është gjithashtu një bartës i shëndetshëm (ose i sëmurë), mund të lindin fëmijë me anemi mesdhetare: ka 25% mundësi që fëmija të zhvillojë një formë të rëndë, një tjetër e barabartë me 25% që të mos jetë i sëmurë dhe 50 të mbetura. % që tregojnë një pamje të lehtë.
Simptomat dhe komplikimet
Anemia mesdhetare shfaqet me pamje klinike shumë të ndryshueshme, të cilat varen nga ashpërsia e aberracionit gjenetik themelor.
Format e lehta të këtij çrregullimi hematologjik mund të jenë pothuajse asimptomatike.
Simptomat janë veçanërisht të rënda në aneminë kryesore mesdhetare dhe mund të përfshijnë:
zbehje progresive;
Lodhje;
Dobësi muskulore;
Zhvillimi i ngadaltë dhe verdhëza tek fëmija;
Ndryshimet e kockave (trashje e kafkës, mollëza të zgjatura, gjuri valgus dhe fraktura patologjike të kockave të gjata);
Anoreksi (mungesë oreksi);
Prishja e kushteve të përgjithshme;
Periudhat e përsëritura të temperaturës së ulët;
Diarre;
Irritimi;
Distension progresiv i barkut (dytësor ndaj splenomegalisë dhe hepatomegalisë).
Me transfuzione të rregullta, rritja dhe zhvillimi i fëmijëve me anemi mesdhetare priren të jenë normale. Megjithatë, me kalimin e kohës, mund të shfaqen komplikime të mbingarkesës me hekur, si zhvillimi i vonuar i trupit dhe maturimi seksual, hemokromatoza dhe sideroza e mëlçisë.
Anemia mesdhetare predispozon zhvillimin e patologjive të ndryshme.
Së pari, për të kompensuar mungesën e qelizave të kuqe të gjakut, palca e eshtrave do të përpiqet të prodhojë më shumë. Ky reagim rezulton në një tendencë për të formuar kocka më të gjata dhe më të brishta, me rrezikun e zhvillimit të osteoporozës. Një mekanizëm i ngjashëm ka të bëjë me shpretkën, e cila do t’i nënshtrohet të njëjtës përpjekje. Pasojat janë një zmadhim i organit i cili për më tepër shpërqendrohet nga funksionet e tjera të tij, si ai i kontributit në mbrojtjen e organizmit nga infeksionet.
Transfuzionet e shpeshta të gjakut, të parashikuara në trajtimin e anemisë së rëndë mesdhetare, krijojnë një akumulim hekuri, me rrezikun e një toksikoze që mund të dëmtojë organe, si mëlçia dhe zemra.
Ndër pasojat e anemisë mesdhetare, mund të vërehen edhe këto:
Çrregullime endokrine dhe çekuilibër hormonal (përfshirë hipotiroidizmin, pamjaftueshmërinë e paratiroideve dhe veshkave dhe diabetin mellitus);
Kolelitiaza (si në sëmundjen drapërocitare);
Ulçera kutane e gjymtyrëve të poshtme për shkak të insuficiencës kronike venoze.
Nga ana tjetër, njerëzit me anemi mesdhetare janë më të mbrojtur kundër malaries: meqenëse qelizat e kuqe të gjakut janë të vogla dhe të pakta, plazmodiumi nuk është në gjendje të shfrytëzojë sistemin e gjakut që do të donte ta parazitonte në avantazhin e tij.
Diagnoza
Nëse diagnostikohet në kohë, anemia mesdhetare nuk ju pengon të bëni një jetë pothuajse normale.
Dyshimi për anemi mesdhetare mund të lindë me një vizitë pas lindjes, për shkak të shfaqjes së simptomave sugjestive (të tilla si verdhëza dhe rritja e dobët).
Përveç simptomave, diagnoza bazohet në disa teste gjaku për të përcaktuar:
sasia dhe lloji i hemoglobinës;
Numri dhe vëllimi i qelizave të kuqe të gjakut.
Më pas diagnoza konfirmohet me anë të testeve gjenetike. Përveç kësaj, konstatohen shenja hemolitike të shoqëruara me eritropoezë joefektive, me hiperbilirubinemi indirekte, hipersideremi dhe hiperferritinemi.
Diagnoza prenatale
Gjatë shtatzënisë është e mundur të verifikohet nëse fëmija do të lindë me anemi mesdhetare, me anë të diagnozës prenatale në vilin korionik ose amniocentezës. Ekzaminimi prenatal është veçanërisht i rëndësishëm në zonat më të përhapura.
Për prindërit e ardhshëm që duan të dinë nëse janë bartës të shëndetshëm të anemisë mesdhetare, është e mundur t’i nënshtrohen një studimi të hemoglobinës, i cili kërkon një mostër të thjeshtë gjaku.
Terapia
Trajtimi përfshin disa qasje, duke përfshirë:
Transfuzionet e gjakut për të kompensuar mungesën e rruazave të kuqe të gjakut, ndoshta të lidhura me terapinë e kelimit për të shmangur akumulimin e hekurit;
Splenektomia (nëse sëmundja shkakton anemi të rëndë ose splenomegali);
Transplantimi i palcës së eshtrave ose qelizave staminale nga donatorë të pajtueshëm.
Në përgjithësi, anemia madhore mesdhetare parashikon trajtim me transfuzione të rregullta gjaku (mesatarisht një çdo 15 ditë në rastet më të rënda), ndërsa në talaseminë intermedia, mjaftojnë disa cikle kur hemoglobina është shumë e ulët.
Megjithatë, transfuzionet shkaktojnë një akumulim të hekurit në trup, i cili mund të shkaktojë probleme veçanërisht në zemër dhe mëlçi. Për të shmangur këto dëmtime, është e nevojshme vendosja e një terapie të vazhdueshme me medikamente chelating hekuri, pra barna të afta për të kapur dhe eliminuar hekurin e tepërt nga trupi.
Në disa raste mund të përdoret transplanti i palcës kockore, e vetmja terapi “definitive” për aneminë mesdhetare. Megjithatë, shanset për të gjetur një donator të pajtueshëm janë të kufizuara (afërsisht 25%), pa marrë parasysh kundërindikacionet dhe rreziqet e këtij lloji të operacionit, përfshirë refuzimin nga trupi. Për këto arsye, kjo procedurë zakonisht rezervohet për raste të rralla, në prani të personave të prekur nga një formë shumë e rëndë e anemisë mesdhetare dhe me donatorë familjarë identikë HLA (vëllezërit ose motrat e pacientit).
Përveç terapive specifike, rëndësi të madhe kanë aktiviteti fizik dhe ushqimi i rregullt.
Në veçanti, mund të jetë e dobishme:
Kufizoni ushqimet me origjinë shtazore dhe të pasura me hekur, si mëlçia, derri, viçi, të brendshmet dhe midhjet;
Shoqëroni vaktet me çaj, në mënyrë që të reduktoni përthithjen e hekurit;
Konsumoni ushqime të pasura me kalcium dhe vitaminë D, për rrezikun e osteoporozës;
Merrni suplemente të acidit folik (për të rritur prodhimin e qelizave të kuqe të gjakut).
Në çdo rast, mjeku do të jetë në gjendje të këshillojë pacientin talasemik për ndërhyrjet më të mira për të menaxhuar gjendjen e tij.
Prognoza
Vëmendja e duhur ndaj aktivitetit fizik dhe të ushqyerit, së bashku me terapinë më të përshtatshme, mund të përmirësojë ndjeshëm cilësinë e jetës së personave që vuajnë nga anemia mesdhetare